If you use this service, could you please send us a mail to npsang@ibcp.fr with details about your usage of the NPSA service (tools used, frequency, type of sequence, ..) ?
Could you explain what makes this service unique for you ?
Could you please add information about your country and your laboratory ?
Thanks
FASTA help
A brief introduction to FASTA
FASTA algorithm is an improvment of FASTP alogithm described in 1985. It allows the comparison of one sequence against a database of one
or several sequences in order to found similar sequences to the query one. The sequences can be protein or DNA ones. The speed and the
sensitivity of the search depend on the word size (ktup) used in the first step of the algorithm.
So, for example, you can use FASTA to find similar sequences in a database built
from an ACNUC query or
after a PATTINPROT search.
Parameters
FASTA parameters are not currently available for the user.
By default the number of description and the number of alignment are set to 500.
The word size (ktup) is set to 2 (less sensitive but faster).
The expected threshold is set to 10.0.
The comparison matrix is BLOSUM50.
The gap opening penalty is -12 and gap extending penalty is -2.
To retrieve these informations see the line "Information and statistics : [FASTA]" in
NPS@ FASTA result file.
NPS@ FASTA output example
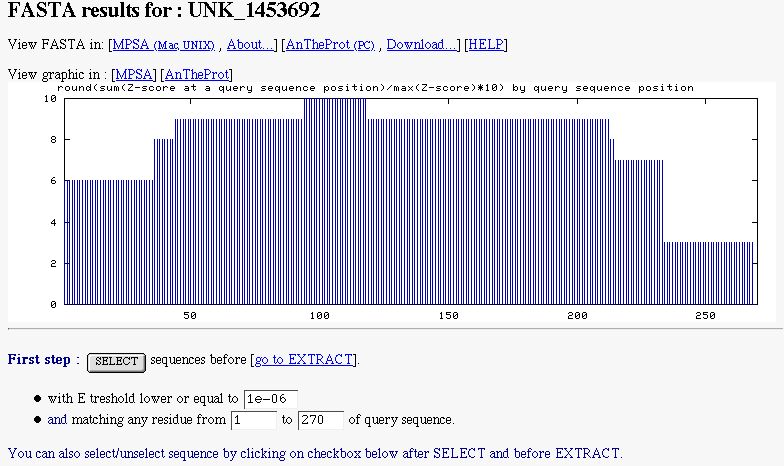
The NPS@ FASTA output is divided into three parts.
A graphic indicating the similarity found along the query sequence. It's computed with FASTA alignment. This graphic can
be see in MPSA/ANTHEPROT to determine interesting boundaries in the query sequence.
A form to select subject sequences. You can select sequences by indicated the maximal E threshold to do it (by default
NPS@ sets it to 1e-6). You can also select subject sequences matching a particular region of the
query.
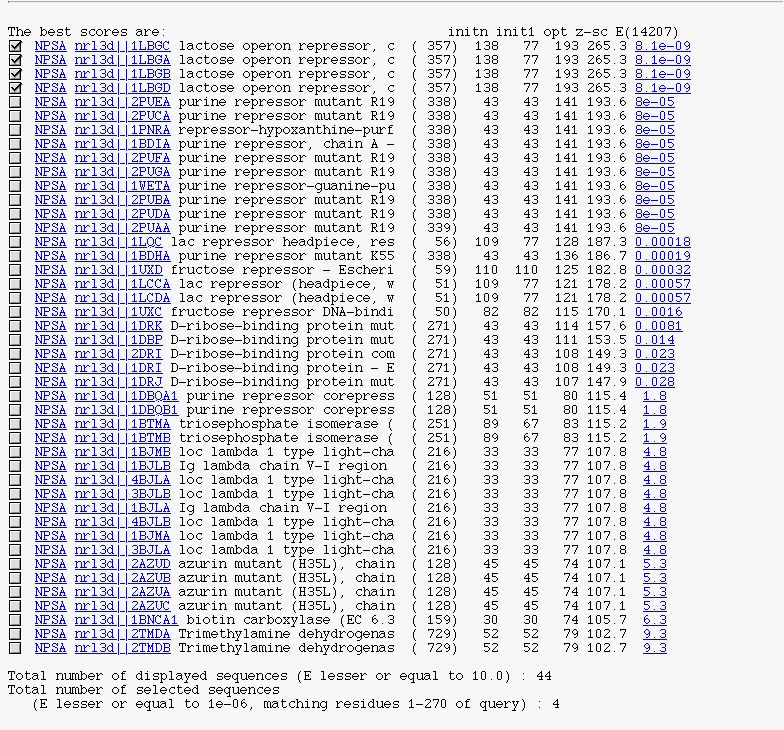
PART 2:
It's the FASTA description block in a 'HTMLized' form.
You can see :
A checkbox to select/unselect subject sequence.
The NPSA link allows you to apply
NPS@ methods on the corresponding sequence after it is extracted from the query database.
The database link to retrieve the database entry.
The alignment link (on expected value) to see FASTA alignment between the query sequence and the current subject sequence.
The number of displayed sequences.
The number of selected sequences.
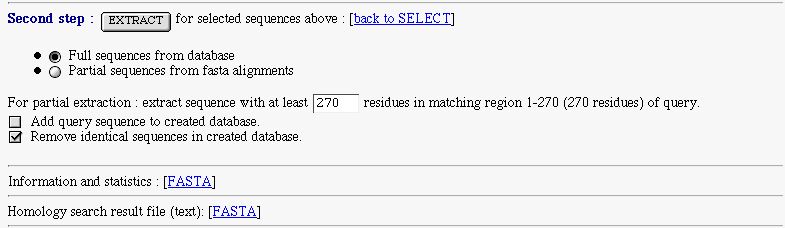
PART 3:
You have :
An extract form to extract selected sequence and make a database. You can extract full or partial sequences.
The full extract is made from the database.
The partial extract, is made for subject sequence that match the query one in the indicated region. The extracted sequence
is retrieved from the FASTA alignment. You can set the minimal length of the extracted sequence.

 A brief introduction to FASTA
A brief introduction to FASTA